Alpha-mannosidosis: Difference between revisions
CSV import |
CSV import |
||
| Line 47: | Line 47: | ||
[[Category:Lysosomal storage disorders]] | [[Category:Lysosomal storage disorders]] | ||
[[Category:Rare diseases]] | [[Category:Rare diseases]] | ||
== Alpha-mannosidosis == | |||
<gallery> | |||
File:autorecessive.svg|Autosomal recessive inheritance pattern | |||
File:Alpha-mannosidosis.JPEG|Alpha-mannosidosis | |||
File:Alpha-mannosidosis electron micrograph.JPEG|Alpha-mannosidosis electron micrograph | |||
</gallery> | |||
Latest revision as of 00:47, 20 February 2025
| Alpha-mannosidosis | |
|---|---|
| Synonyms | N/A |
| Pronounce | N/A |
| Specialty | N/A |
| Symptoms | Intellectual disability, hearing loss, skeletal abnormalities |
| Complications | N/A |
| Onset | Childhood |
| Duration | Lifelong |
| Types | N/A |
| Causes | Genetic mutation in the MAN2B1 gene |
| Risks | N/A |
| Diagnosis | Genetic testing, clinical evaluation |
| Differential diagnosis | N/A |
| Prevention | N/A |
| Treatment | Enzyme replacement therapy, hematopoietic stem cell transplantation |
| Medication | N/A |
| Prognosis | Variable |
| Frequency | Rare |
| Deaths | N/A |
Alpha-mannosidosis is a rare genetic disorder characterized by the body's inability to properly break down certain complex sugars due to a deficiency of the enzyme alpha-mannosidase. This condition is classified as a lysosomal storage disorder.
Signs and Symptoms[edit]
Individuals with alpha-mannosidosis may exhibit a range of symptoms, including intellectual disability, hearing loss, and skeletal abnormalities. Other common features include immune system deficiencies, facial dysmorphism, and motor function impairment. Symptoms typically appear in childhood and can vary in severity.
Causes[edit]
Alpha-mannosidosis is caused by mutations in the MAN2B1 gene, which provides instructions for producing the enzyme alpha-mannosidase. This enzyme is essential for the breakdown of oligosaccharides in the lysosome. Mutations in the MAN2B1 gene lead to a deficiency of this enzyme, resulting in the accumulation of undigested sugars in the body's cells.
Diagnosis[edit]
Diagnosis of alpha-mannosidosis is based on clinical evaluation and confirmed through genetic testing. Biochemical tests may also be used to measure the activity of the alpha-mannosidase enzyme in blood or tissue samples.
Treatment[edit]
Currently, treatment options for alpha-mannosidosis include enzyme replacement therapy and hematopoietic stem cell transplantation. These treatments aim to reduce symptoms and improve quality of life. Supportive therapies, such as physical therapy and speech therapy, may also be beneficial.
Prognosis[edit]
The prognosis for individuals with alpha-mannosidosis varies depending on the severity of the condition and the effectiveness of treatment. Early diagnosis and intervention can improve outcomes and quality of life.
See Also[edit]
References[edit]
<references/>
External Links[edit]
Alpha-mannosidosis[edit]
-
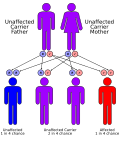 Autosomal recessive inheritance pattern
Autosomal recessive inheritance pattern -
 Alpha-mannosidosis
Alpha-mannosidosis -
 Alpha-mannosidosis electron micrograph
Alpha-mannosidosis electron micrograph



