Fabry disease: Difference between revisions
CSV import |
CSV import |
||
| Line 48: | Line 48: | ||
[[Category:Lysosomal storage diseases]] | [[Category:Lysosomal storage diseases]] | ||
[[Category:Rare diseases]] | [[Category:Rare diseases]] | ||
<gallery> | |||
File:PBB Protein GLA image.jpg|Fabry disease | |||
File:Morbus Fabry Cornea verticillata 01.jpg|Fabry disease | |||
File:Angiokreatoma.jpg|Fabry disease | |||
</gallery> | |||
Revision as of 01:20, 20 February 2025
Fabry Disease
Fabry disease is a rare genetic disorder that results from the buildup of a particular type of fat, called globotriaosylceramide, in the body's cells. It is an X-linked recessive disorder caused by mutations in the GLA gene, which leads to a deficiency of the enzyme alpha-galactosidase A.
Pathophysiology
Fabry disease is characterized by the accumulation of glycosphingolipids in the lysosomes of various cell types. The deficiency of alpha-galactosidase A enzyme activity results in the progressive deposition of globotriaosylceramide in the vascular endothelium, smooth muscle cells, and other tissues, leading to multi-systemic manifestations.
Clinical Manifestations
The symptoms of Fabry disease can vary widely among affected individuals, but common manifestations include:
- Angiokeratomas: Small, dark red spots on the skin, often found in the "bathing trunk" area.
- Acroparesthesia: Episodes of pain and burning sensations in the hands and feet.
- Corneal verticillata: Whorled patterns on the cornea, detectable by an eye examination.
- Renal impairment: Progressive kidney damage leading to chronic kidney disease.
- Cardiac involvement: Hypertrophic cardiomyopathy, arrhythmias, and heart failure.
- Cerebrovascular disease: Increased risk of stroke and transient ischemic attacks.
Diagnosis
Diagnosis of Fabry disease is typically confirmed through:
- Enzyme assay: Measurement of alpha-galactosidase A activity in blood or skin fibroblasts.
- Genetic testing: Identification of mutations in the GLA gene.
- Biopsy: Histological examination of affected tissues may show characteristic lipid deposits.
Treatment
The management of Fabry disease includes:
- Enzyme replacement therapy (ERT): Intravenous administration of recombinant alpha-galactosidase A to reduce globotriaosylceramide accumulation.
- Chaperone therapy: Use of pharmacological chaperones to stabilize the mutant enzyme and enhance its activity.
- Symptomatic treatment: Pain management, renal protection, and cardiovascular care.
- Gene therapy: An emerging approach aimed at correcting the underlying genetic defect.
Prognosis
The prognosis of Fabry disease varies depending on the severity of organ involvement and the effectiveness of treatment. Early diagnosis and initiation of therapy can improve outcomes and quality of life.
Epidemiology
Fabry disease is estimated to affect approximately 1 in 40,000 to 1 in 117,000 live births. It is more common in males due to its X-linked inheritance pattern, but females can also be affected, often with milder symptoms.
See Also
Template:Lysosomal storage disorders
| Genetic disorders relating to deficiencies of transcription factor or coregulators | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
-
 Fabry disease
Fabry disease -
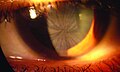 Fabry disease
Fabry disease -
 Fabry disease
Fabry disease



