Tyrosinemia type I
A rare genetic disorder affecting tyrosine metabolism
Tyrosinemia type I is a rare autosomal recessive genetic disorder that affects the metabolism of the amino acid tyrosine. It is caused by a deficiency of the enzyme fumarylacetoacetate hydrolase (FAH), which is the last enzyme in the tyrosine catabolic pathway. This deficiency leads to the accumulation of toxic metabolites, causing severe liver and kidney dysfunction.
Pathophysiology

In Tyrosinemia type I, the deficiency of FAH results in the accumulation of fumarylacetoacetate, which is converted to succinylacetone. Succinylacetone is a toxic compound that inhibits porphobilinogen synthase, leading to porphyria-like symptoms. The accumulation of these metabolites causes damage to the liver and kidneys, leading to hepatorenal syndrome.
Genetics

Tyrosinemia type I is inherited in an autosomal recessive manner. This means that an affected individual must inherit two copies of the defective gene, one from each parent. The gene responsible for Tyrosinemia type I is located on chromosome 15q23-q25 and is known as the FAH gene.
Symptoms
Symptoms of Tyrosinemia type I typically appear in infancy and may include failure to thrive, jaundice, hepatomegaly, renal tubular acidosis, and rickets. If untreated, the condition can lead to liver failure, renal failure, and an increased risk of hepatocellular carcinoma.
Diagnosis
Diagnosis of Tyrosinemia type I is based on clinical symptoms, biochemical tests showing elevated levels of tyrosine and succinylacetone in the blood and urine, and genetic testing to confirm mutations in the FAH gene.
Treatment

The primary treatment for Tyrosinemia type I is the use of nitisinone, a drug that inhibits the enzyme 4-hydroxyphenylpyruvate dioxygenase, thereby reducing the production of toxic metabolites. Dietary management with a low-tyrosine and low-phenylalanine diet is also essential. In severe cases, liver transplantation may be necessary.
Prognosis
With early diagnosis and treatment, individuals with Tyrosinemia type I can have a significantly improved prognosis. Nitisinone therapy has been shown to prevent liver and kidney damage and reduce the risk of liver cancer.
Related pages
References
- Grompe, M. (2001). "The pathophysiology and treatment of hereditary tyrosinemia type 1." Seminars in Liver Disease, 21(4), 563-571.
- Lindstedt, S., et al. (1992). "Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase." The Lancet, 340(8823), 813-817.
-
 Tyrosinemia type I metabolic pathway
Tyrosinemia type I metabolic pathway -
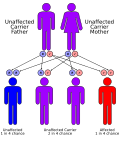 Autosomal recessive inheritance pattern
Autosomal recessive inheritance pattern -
 Tyrosine metabolic pathway
Tyrosine metabolic pathway -
 Tyrosine WPMP
Tyrosine WPMP -
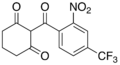 Nitisinone structure
Nitisinone structure


Ad. Transform your life with W8MD's
GLP-1 weight loss injections special from $29.99 with insurance
|
WikiMD Medical Encyclopedia |
Medical Disclaimer: WikiMD is for informational purposes only and is not a substitute for professional medical advice. Content may be inaccurate or outdated and should not be used for diagnosis or treatment. Always consult your healthcare provider for medical decisions. Verify information with trusted sources such as CDC.gov and NIH.gov. By using this site, you agree that WikiMD is not liable for any outcomes related to its content. See full disclaimer.
Credits:Most images are courtesy of Wikimedia commons, and templates, categories Wikipedia, licensed under CC BY SA or similar.
Translate this page: - East Asian
中文,
日本,
한국어,
South Asian
हिन्दी,
தமிழ்,
తెలుగు,
Urdu,
ಕನ್ನಡ,
Southeast Asian
Indonesian,
Vietnamese,
Thai,
မြန်မာဘာသာ,
বাংলা
European
español,
Deutsch,
français,
Greek,
português do Brasil,
polski,
română,
русский,
Nederlands,
norsk,
svenska,
suomi,
Italian
Middle Eastern & African
عربى,
Turkish,
Persian,
Hebrew,
Afrikaans,
isiZulu,
Kiswahili,
Other
Bulgarian,
Hungarian,
Czech,
Swedish,
മലയാളം,
मराठी,
ਪੰਜਾਬੀ,
ગુજરાતી,
Portuguese,
Ukrainian
