Usual interstitial pneumonia: Difference between revisions
CSV import |
CSV import Tags: mobile edit mobile web edit |
||
| Line 33: | Line 33: | ||
[[Category:Interstitial lung diseases]] | [[Category:Interstitial lung diseases]] | ||
[[Category:Pulmonary fibrosis]] | [[Category:Pulmonary fibrosis]] | ||
<gallery> | |||
File:CT_scan_in_usual_interstitial_pneumonia_(UIP).jpg|CT scan showing usual interstitial pneumonia | |||
File:UIP_(Usual_interstitial_pneumonia)-CT_scan.jpg|CT scan of usual interstitial pneumonia | |||
File:UIPlungbiopsy.jpg|Lung biopsy showing usual interstitial pneumonia | |||
File:Honeycomb_change.jpg|Honeycomb change in usual interstitial pneumonia | |||
File:Fibroblast_focus.jpg|Fibroblast focus in usual interstitial pneumonia | |||
</gallery> | |||
Revision as of 05:00, 18 February 2025
Usual Interstitial Pneumonia
.jpg)
-CT_scan.jpg)



Usual interstitial pneumonia (UIP) is a specific form of lung disease characterized by progressive scarring of both lungs. It is the most common form of idiopathic pulmonary fibrosis (IPF), a type of interstitial lung disease.
Clinical Features
Patients with UIP typically present with dyspnea (shortness of breath) and a persistent dry cough. The disease is more common in older adults, particularly those over the age of 50. Physical examination may reveal clubbing of the fingers and crackles on lung auscultation.
Pathology
The hallmark of UIP is the presence of patchy interstitial fibrosis, which is most pronounced in the subpleural and basal regions of the lungs. Histologically, UIP is characterized by the presence of fibroblastic foci, honeycomb change, and temporal heterogeneity of fibrosis.
Diagnosis
Diagnosis of UIP is typically made through a combination of clinical, radiological, and pathological findings. High-resolution computed tomography (HRCT) scans of the chest are crucial for diagnosis, often showing a pattern of reticular opacities, traction bronchiectasis, and honeycombing. A lung biopsy may be performed to confirm the diagnosis.
Treatment
There is no cure for UIP, and treatment is primarily supportive. Antifibrotic medications such as pirfenidone and nintedanib may slow disease progression. Lung transplantation is an option for eligible patients with advanced disease.
Prognosis
The prognosis for patients with UIP is generally poor, with a median survival of 3 to 5 years after diagnosis. The disease course is variable, with some patients experiencing rapid progression and others having a more indolent course.
Related Pages
| Pulmonology topics | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
This pulmonology-related article is a stub. You can help WikiMD by expanding it.
|
-
 CT scan showing usual interstitial pneumonia
CT scan showing usual interstitial pneumonia -
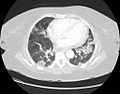 CT scan of usual interstitial pneumonia
CT scan of usual interstitial pneumonia -
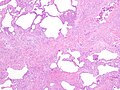 Lung biopsy showing usual interstitial pneumonia
Lung biopsy showing usual interstitial pneumonia -
 Honeycomb change in usual interstitial pneumonia
Honeycomb change in usual interstitial pneumonia -
 Fibroblast focus in usual interstitial pneumonia
Fibroblast focus in usual interstitial pneumonia
