Computational chemistry: Difference between revisions
CSV import |
CSV import |
||
| Line 39: | Line 39: | ||
{{Chemistry-stub}} | {{Chemistry-stub}} | ||
== Computational_chemistry == | |||
<gallery> | |||
File:C60_isosurface.png|C60 isosurface | |||
File:CatalysisScheme.png|Catalysis Scheme | |||
File:Electron_correlation.svg|Electron correlation | |||
File:Diazomethane-pi-system.png|Diazomethane pi system | |||
File:MM_PEF_3.png|MM PEF 3 | |||
File:A_molecular_dynamics_simulation_of_argon_gas.webm|A molecular dynamics simulation of argon gas | |||
File:A_Molecular_Dynamics_Simulation_of_Liquid_Water_at_298_K.webm|A Molecular Dynamics Simulation of Liquid Water at 298 K | |||
File:Hartree-Fock.png|Hartree-Fock | |||
File:Acrolein-s-trans-GED-MW-3D-sf.png|Acrolein s-trans GED MW 3D sf | |||
File:Methan_2a1.png|Methan 2a1 | |||
</gallery> | |||
Latest revision as of 21:43, 23 February 2025
Computational chemistry is an interdisciplinary field that uses computer simulation to assist in solving chemical problems. It uses methods of theoretical chemistry, incorporated into efficient computer programs, to calculate the structures and properties of molecules and solids. It is necessary because, apart from relatively recent results concerning the hydrogen molecular ion (dihydrogen cation), the quantum n-body problem cannot be solved analytically, much less in closed form. Computational chemistry is therefore often used when a mathematical solution is impractical or when a large number of calculations are required, such as in the study of macromolecules.
Overview[edit]
Computational chemistry can be divided into the study of ab initio and semi-empirical methods. The former involves methods that aim to solve the Schrödinger equation directly with no approximations (the term ab initio is Latin for 'from the beginning'), while the latter involves methods that incorporate empirical parameters derived from experimental data to simplify the calculations. Another important aspect of computational chemistry is molecular dynamics, which simulates the physical movements of atoms and molecules over time, and quantum mechanics, a fundamental theory in physics that provides a description of the physical properties of nature at the scale of atoms and subatomic particles.
Applications[edit]
Computational chemistry has a wide range of applications, including drug design, where it is used to predict the interaction between molecules and biological targets; materials science, where it helps in the design of new materials with desired properties; and environmental chemistry, where it is used to model the behavior of pollutants and predict their effects on the environment. It also plays a crucial role in understanding fundamental aspects of chemistry, such as reaction mechanisms and the nature of chemical bonds.
Methods[edit]
Ab Initio Methods[edit]
Ab initio methods, such as Hartree-Fock (HF) and post-Hartree-Fock, aim to solve the Schrödinger equation without any empirical input. These methods are highly accurate but computationally demanding, making them suitable for small molecules.
Semi-Empirical Methods[edit]
Semi-empirical methods, including AM1, PM3, and MNDO, use empirical parameters to simplify calculations. These methods are less accurate than ab initio methods but are much faster, making them suitable for larger molecules.
Density Functional Theory (DFT)[edit]
Density Functional Theory (DFT) is a computational quantum mechanical modelling method used in physics, chemistry, and materials science to investigate the electronic structure (principally the ground state) of many-body systems, especially atoms, molecules, and the condensed phases. DFT has become very popular for chemical calculations due to its favorable balance between accuracy and computational cost.
Molecular Dynamics[edit]
Molecular dynamics (MD) simulations use classical mechanics to model the behavior of atoms and molecules over time. MD is particularly useful for studying the physical properties of matter, such as diffusion and conformational changes in biomolecules.
Software[edit]
Several software packages are available for computational chemistry, including Gaussian, VASP, ABINIT, and NWChem. These packages vary in their capabilities, with some focusing on specific types of calculations and others offering a broad range of functionalities.
Challenges and Future Directions[edit]
Despite its successes, computational chemistry faces challenges, such as the need for more accurate and efficient methods, the handling of large and complex systems, and the integration of quantum mechanics and molecular dynamics simulations. Ongoing research in the field aims to address these challenges and expand the applicability of computational chemistry to new areas of research.
See Also[edit]
Computational_chemistry[edit]
-
 C60 isosurface
C60 isosurface -
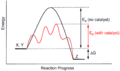 Catalysis Scheme
Catalysis Scheme -
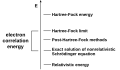 Electron correlation
Electron correlation -
 Diazomethane pi system
Diazomethane pi system -
 MM PEF 3
MM PEF 3 -
 A molecular dynamics simulation of argon gas
A molecular dynamics simulation of argon gas -
A Molecular Dynamics Simulation of Liquid Water at 298 K
-
 Hartree-Fock
Hartree-Fock -
 Acrolein s-trans GED MW 3D sf
Acrolein s-trans GED MW 3D sf -
 Methan 2a1
Methan 2a1








