Hunter syndrome: Difference between revisions
CSV import |
CSV import |
||
| Line 43: | Line 43: | ||
[[Category:Lysosomal storage disorders]] | [[Category:Lysosomal storage disorders]] | ||
[[Category:Rare diseases]] | [[Category:Rare diseases]] | ||
<gallery> | |||
File:Mucopolysaccharidosis (Hunter's Syndrome) 3.jpg|Hunter syndrome | |||
File:X-linked recessive.svg|X-linked recessive inheritance | |||
File:Dermatan sulfate.svg|Dermatan sulfate | |||
</gallery> | |||
Revision as of 01:06, 20 February 2025
A rare genetic disorder affecting metabolism
Hunter syndrome, also known as mucopolysaccharidosis type II (MPS II), is a rare genetic disorder that primarily affects males. It is one of the lysosomal storage disorders and is caused by a deficiency of the enzyme iduronate-2-sulfatase. This enzyme deficiency leads to the accumulation of glycosaminoglycans (GAGs) in the body's cells, which can cause a variety of symptoms and complications.
Genetics
Hunter syndrome is an X-linked recessive disorder, meaning the defective gene is located on the X chromosome. Since males have only one X chromosome, a single defective gene is sufficient to cause the disorder. Females, having two X chromosomes, are typically carriers and usually do not exhibit symptoms, although some may have mild manifestations.
Pathophysiology
The enzyme iduronate-2-sulfatase is responsible for breaking down complex molecules known as glycosaminoglycans. In individuals with Hunter syndrome, the deficiency of this enzyme leads to the accumulation of GAGs, such as dermatan sulfate and heparan sulfate, in various tissues. This accumulation disrupts normal cellular function and leads to the symptoms associated with the disorder.
Symptoms
The symptoms of Hunter syndrome can vary widely among affected individuals and may include:
- Developmental delay
- Coarse facial features
- Enlarged liver and spleen (hepatosplenomegaly)
- Joint stiffness
- Hearing loss
- Heart valve abnormalities
- Respiratory issues
Symptoms typically appear between the ages of 2 and 4 years and can progress over time. The severity of the disorder can range from mild to severe.
Diagnosis
Diagnosis of Hunter syndrome is based on clinical evaluation, family history, and laboratory tests. Enzyme assays can measure the activity of iduronate-2-sulfatase in blood or skin cells. Genetic testing can confirm the diagnosis by identifying mutations in the IDS gene.
Treatment
Currently, there is no cure for Hunter syndrome, but treatments are available to manage symptoms and improve quality of life. These include:
- Enzyme replacement therapy (ERT) with idursulfase, which can help reduce GAG accumulation.
- Supportive care, such as physical therapy, occupational therapy, and speech therapy.
- Surgical interventions for complications like carpal tunnel syndrome or heart valve issues.
Prognosis
The prognosis for individuals with Hunter syndrome varies depending on the severity of the condition. Those with a milder form may live into adulthood, while those with a more severe form may have a reduced life expectancy due to complications such as respiratory failure or cardiac issues.
Related pages
-
 Hunter syndrome
Hunter syndrome -
 X-linked recessive inheritance
X-linked recessive inheritance -
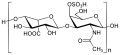 Dermatan sulfate
Dermatan sulfate
_3.jpg)


