Acute interstitial pneumonitis: Difference between revisions
CSV import |
CSV import |
||
| Line 36: | Line 36: | ||
[[Category:Respiratory diseases]] | [[Category:Respiratory diseases]] | ||
[[Category:Interstitial lung diseases]] | [[Category:Interstitial lung diseases]] | ||
<gallery> | |||
File:Hyaline_membranes_-_very_high_mag.jpg|Hyaline membranes under very high magnification | |||
File:CRIM.PULMONOLOGY2012-678249.001.jpg|Acute interstitial pneumonia (AIP) histological image | |||
File:Acute_interstitial_pneumonia_(AIP)_Idiopathic_DAD_3.jpg|Acute interstitial pneumonia (AIP) Idiopathic DAD | |||
File:Acute_interstitial_pneumonia_(AIP)_Idiopathic_DAD_2.jpg|Acute interstitial pneumonia (AIP) Idiopathic DAD | |||
File:Acute_interstitial_pneumonia_(AIP)_Idiopathic_DAD.jpg|Acute interstitial pneumonia (AIP) Idiopathic DAD | |||
</gallery> | |||
Revision as of 11:27, 18 February 2025
Acute interstitial pneumonitis


Acute interstitial pneumonitis (AIP), also known as Hamman-Rich syndrome, is a rare and severe form of interstitial lung disease characterized by the rapid onset of respiratory failure. It is classified as an idiopathic form of diffuse alveolar damage (DAD) and is considered a type of acute respiratory distress syndrome (ARDS).
Pathophysiology
AIP is characterized by diffuse alveolar damage, which involves injury to the alveolar epithelium and capillary endothelium. This leads to the formation of hyaline membranes, interstitial edema, and infiltration of inflammatory cells. The exact cause of AIP is unknown, but it is thought to be triggered by an acute inflammatory response in the lungs.
Clinical presentation
Patients with AIP typically present with sudden onset of dyspnea, cough, and fever. The condition progresses rapidly to severe respiratory failure, often requiring mechanical ventilation. The clinical course is similar to that of ARDS, but AIP is distinguished by its idiopathic nature and histological findings.
Diagnosis
The diagnosis of AIP is based on clinical presentation, imaging studies, and histopathological examination. High-resolution computed tomography (HRCT) of the chest often shows diffuse bilateral ground-glass opacities and consolidation. A lung biopsy is usually required to confirm the diagnosis, revealing the characteristic diffuse alveolar damage with hyaline membrane formation.
Treatment
There is no specific treatment for AIP, and management is primarily supportive. Patients often require mechanical ventilation and intensive care. Corticosteroids and other immunosuppressive agents may be used, but their efficacy is uncertain. The prognosis is generally poor, with a high mortality rate.
Prognosis
The prognosis of AIP is poor, with a high mortality rate, often exceeding 50%. Survivors may experience long-term pulmonary sequelae, including pulmonary fibrosis.
Related pages
- Interstitial lung disease
- Acute respiratory distress syndrome
- Diffuse alveolar damage
- Pulmonary fibrosis
Gallery
-
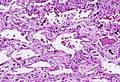 Histopathological image showing diffuse alveolar damage
Histopathological image showing diffuse alveolar damage -
 Another view of diffuse alveolar damage in AIP
Another view of diffuse alveolar damage in AIP -
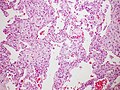 Diffuse alveolar damage with hyaline membranes
Diffuse alveolar damage with hyaline membranes
_Idiopathic_DAD_3.jpg)
_Idiopathic_DAD_2.jpg)
_Idiopathic_DAD.jpg)
-
 Hyaline membranes under very high magnification
Hyaline membranes under very high magnification -
 Acute interstitial pneumonia (AIP) histological image
Acute interstitial pneumonia (AIP) histological image -
Acute interstitial pneumonia (AIP) Idiopathic DAD
-
Acute interstitial pneumonia (AIP) Idiopathic DAD
-
Acute interstitial pneumonia (AIP) Idiopathic DAD
